
Big pharma marketers who’ve been complaining to Washington for decades about the unregulated nibbling at their market shares by counterfeiters and large-scale domestic drug compounders won historic relief when President Obama signed the Drug Quality and Security Act into law on Nov. 27.
As with anything Washington does, the new law is the product of compromise, making it weaker than it could be. Its chief weakness: pharmacy compounder registration with FDA is voluntary, potentially letting rogue operations remain hidden until patient injuries have exposed them.
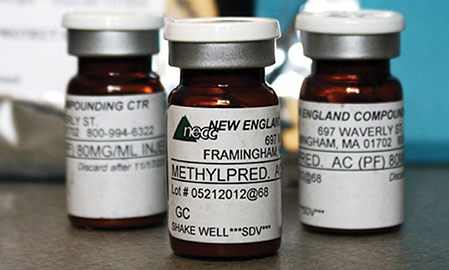
Spurred by this, FDA took just seven days, including the Thanksgiving weekend, to publish six Federal Register notices on compounding. Those included three draft guidance documents based on the new law, under which the agency discussed enforcement strategies intended to prevent a future public health crisis like the 2012 meningitis outbreak tied to the New England Compounding Center (NECC), as well as securing the pharmaceutical supply chain.
NECC shipped its non-sterile, preservative-free versions of methylprednisolone acetate (originally Pfizer’s Depo-Medrol) and triamcinolone acetonide (originally Bristol Myers-Squibb’s Kenalog), among other compromised drugs, to 75 medical facilities in 23 states where they were administered to 14,000 patients, 48 of whom subsequently died.
More than 400 lawsuits have been filed against the company and its owners; it has filed for bankruptcy and is no longer in operation. At least seven other large-scale interstate compounding operations have been cited or shut down by state regulators over similar issues.
Key provisions of the new law restrict FDA-registered compounders (now called “outsourcers”) to FDA-listed or compendially recognized bulk drug substances, prohibit them from making drugs withdrawn from the market due to safety or ineffectiveness reasons; bar them from making drugs that are “essentially a copy of one or more approved drugs”; and exclude them from compounding drugs that FDA has listed as having “demonstrable difficulties for compounding.”
Other provisions establish uniform labeling and packaging requirements, ban the wholesaling of compounded drugs, and require outsourcers to imitate FDA “risk evaluation and mitigation strategies” required of mainstream manufacturers.
Among the flurry of FDA documents, one noted that the agency and the National Association of Boards of Pharmacy (NABP) will need to draft a memorandum of understanding to address “interstate distribution of inordinate amounts of compounded drug products and provide for appropriate investigation by a state agency of complaints relating to compounded drug products distributed outside that state.”
However, FDA’s associate director for drug policy, Jane Axelrad, acknowledged that the law doesn’t give FDA authority to seek out and inspect compounders that elect not to register with FDA.
A second document would require all “outsourcing facilities” to annually report to FDA all drugs compounded during the previous six-month period and provide for each its active ingredient and strength, its source and National Drug Code number, the dosage form and route of administration, the package description, number of individual units produced and NDC number of the final product, if assigned.
In a statement, FDA commissioner Margaret Hamburg wrote that facilities will be subject to inspection by FDA on a risk-based schedule, and her agency “will be encouraging healthcare providers and health networks to consider purchasing compounded products from facilities that are registered with FDA.”
A third FDA document outlined a new drug track-and-trace system that will, for the first time ever, track prescription drugs down to unit level, through standardized serial numbers, from manufacturing to all points of distribution, within a decade.
The law is based on a requirement that all drug products have a unique identifier that manufacturers must maintain for at least six years. It requires FDA to issue a draft guidance document establishing internationally recognized standards “for the interoperable exchange of transaction information, transaction history, and transaction statements,” to lot level, for all members of the supply chain.
Dispensers are banned from accepting products that do not have all three sets of documents. Manufacturers and wholesalers must quarantine products that lack them and report to FDA.
From the January 01, 2014 Issue of MM+M - Medical Marketing and Media





